Metabolic models predict bacterial passengers in colorectal cancer (Cancer & Metabolism)
Metabolic models predict bacterial passengers in colorectal cancer
Daniel R. Garza, Rahwa Taddese, Jakob Wirbel, Georg Zeller, Annemarie Boleij, Martijn A. Huynen & Bas E. Dutilh
Background
Colorectal cancer (CRC) is a complex multifactorial disease. Increasing evidence suggests that the microbiome is involved in different stages of CRC initiation and progression. Beyond specific pro-oncogenic mechanisms found in pathogens, metagenomic studies indicate the existence of a microbiome signature, where particular bacterial taxa are enriched in the metagenomes of CRC patients. Here, we investigate to what extent the abundance of bacterial taxa in CRC metagenomes can be explained by the growth advantage resulting from the presence of specific CRC metabolites in the tumor microenvironment.
Methods
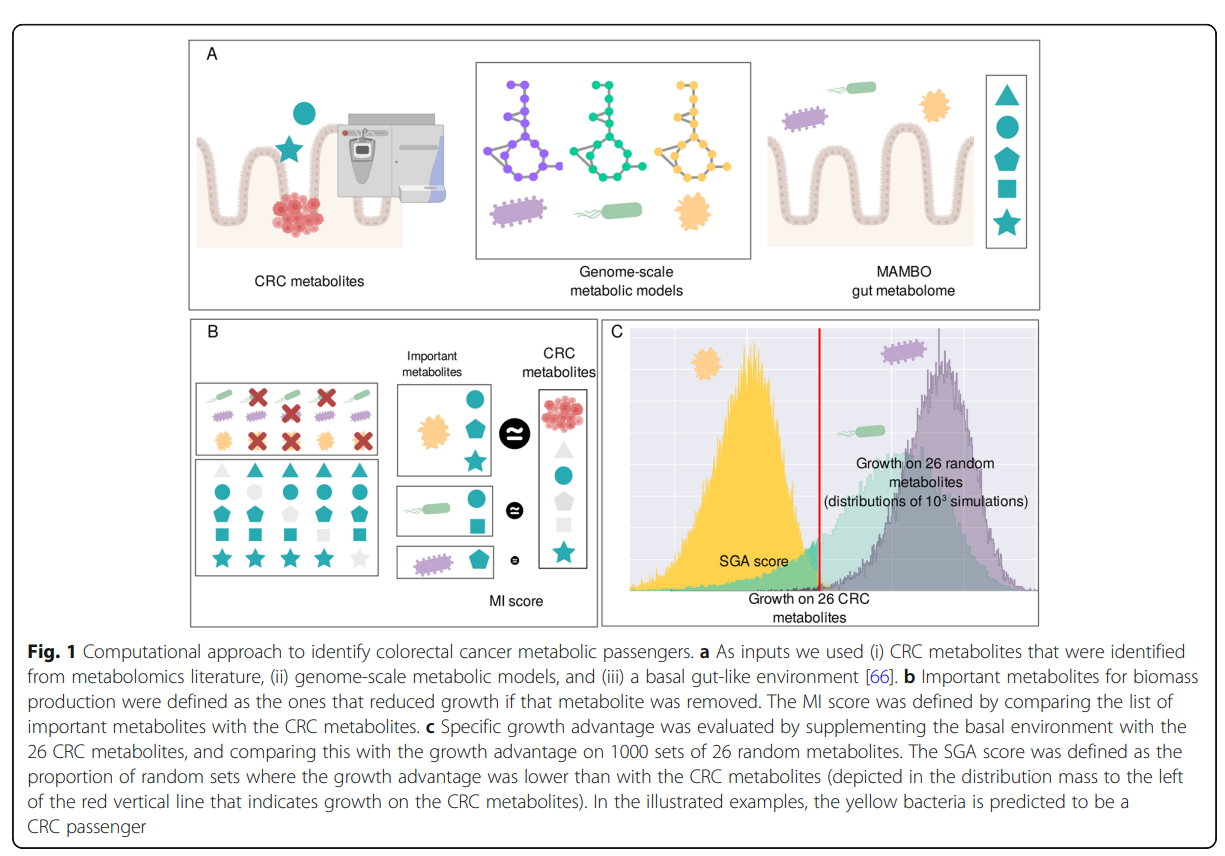
We composed lists of metabolites and bacteria that are enriched on CRC samples by reviewing metabolomics experimental literature and integrating data from metagenomic case-control studies. We computationally evaluated the growth effect of CRC enriched metabolites on over 1500 genome-based metabolic models of human microbiome bacteria. We integrated the metabolomics data and the mechanistic models by using scores that quantify the response of bacterial biomass production to CRC-enriched metabolites and used these scores to rank bacteria as potential CRC passengers.
Results
We found that metabolic networks of bacteria that are significantly enriched in CRC metagenomic samples either depend on metabolites that are more abundant in CRC samples or specifically benefit from these metabolites for biomass production. This suggests that metabolic alterations in the cancer environment are a major component shaping the CRC microbiome.
Conclusion
Here, we show with in sillico models that supplementing the intestinal environment with CRC metabolites specifically predicts the outgrowth of CRC-associated bacteria. We thus mechanistically explain why a range of CRC passenger bacteria are associated with CRC, enhancing our understanding of this disease. Our methods are applicable to other microbial communities, since it allows the systematic investigation of how shifts in the microbiome can be explained from changes in the metabolome.